Molekülarchitekturen aus Atomen modelliert – neuer Vorschlag zur analogen Simulation von Quantenchemie
Physik-News vom 10.10.2019
Ein globales Team an Wissenschaftlern entwickelt die erste Blaupause zur exakten Berechnung molekularer Chemie mittels eines analogen Quantensimulators.Neue Wirkstoffe suchen, neue Verfahren in der chemischen Industrie entwickeln: Computersimulation von Molekülen oder Reaktionen sollen derlei beschleunigen. Doch selbst Supercomputer stoßen dabei schnell an Grenzen.
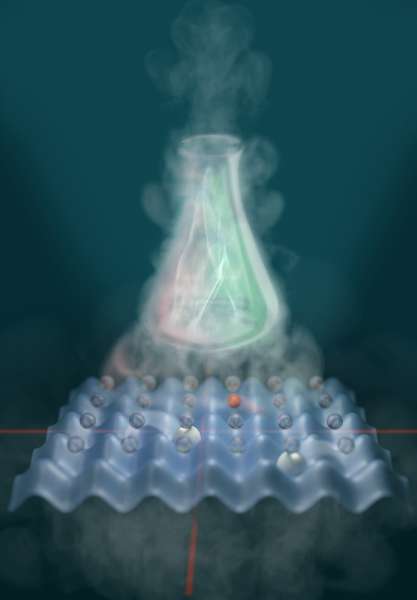
Einen alternativen, analogen Weg zeigen nun Forscher des Max-Planck-Instituts für Quantenoptik in Garching (MPQ). Ein internationales Team um Javier Argüello-Luengo, Doktorand am Institute of Photonic Sciences (ICFO), Ignacio Cirac, Direktor und Leiter der Theorieabteilung am MPQ, Peter Zoller, Direktor am Institut für Quantenoptik und Quanteninformation in Innsbruck (IQOQI), und andere hat nun erstmals eine Blaupause für einen Quantensimulator entworfen, der die Quantenchemie von Molekülen nachahmt. So wie man mit einem Architekturmodell die Statik des späteren Gebäudes prüfen kann, lassen sich mit dem Molekülsimulator Eigenschaften von Molekülen untersuchen. Die Forscher publizierten ihre Ergebnisse im Fachmagazin Nature.
Publikation:
J. Argüello-Luengo, A. Gonzàlez-Tudela, T. Shi, P. Zoller & J. I. Cirac
Analogue Quantum Chemistry Simulation
Nature 574, 215–218 (2019)
DOI: 10.1038/s41586-019-1614-4
Anhand des Beispiels Wasserstoff, dem einfachsten aller Moleküle, demonstrierte das globale Team mit Wissenschaftlern aus Garching, Barcelona, Innsbruck, Madrid und Beijing, wie der Quantensimulator das Verhalten der Elektronenhülle eines realen Moleküls reproduzieren kann. Außerdem zeigen sie in ihrer Arbeit, wie Experimentalphysiker einen solchen Simulator schrittweise aufbauen können. „Unsere Ergebnisse bieten einen neuen Ansatz zur Erforschung der Phänomene in der Quantenchemie“, sagt Javier Argüello-Luengo. Chemiker haben daran großes Interesse, weil Computer sich mit der Simulation von chemischen Verbindungen schwertun. Denn Moleküle gehorchen der Quantenphysik. Ein Elektron in seiner Hülle etwa kann simultan links und rechts herum rotieren. Bei einem Verbund aus vielen Teilchen, wie einem Molekül, potenziert sich die Zahl dieser parallelen Möglichkeiten. Weil jedes Elektron mit jedem anderen wechselwirkt, wächst die Komplexität ins Unermessliche.
Als Ausweg schlug der amerikanische Physiker Richard Feynman 1982 Folgendes vor: Quantensysteme simulieren, indem man sie als vereinfachte Modelle im Labor aus einzelnen Atomen regelrecht nachbaut. Die Parallelität der Möglichkeiten in der Quantenphysik wäre damit automatisch impliziert. Heute sind Quantensimulatoren bereits im Einsatz, sie ahmen zum Beispiel Kristalle nach. Deren regelmäßiges, dreidimensionales Atomgitter imitieren Physiker durch viele sich kreuzende Laserstrahlen, das „optische Gitter“. Die Kreuzungspunkte bilden so etwas wie Mulden in einem Eierkarton, in die Atome gefüllt werden. Durch Verstärken oder Abschwächen der Strahlen kann die Wechselwirkung zwischen den Atomen kontrolliert werden. So erhalten Forscher ein variables Modell, mit dem sie atomares Verhalten exakt studieren können.
Die große Hürde
Neu an dem Vorschlag des internationalen Teams ist die Idee, einen ähnlichen Aufbau für die Simulation eines Moleküls zu nutzen. Die Chemie eines Moleküls wird durch seine Elektronenhülle bestimmt. Im theoretischen Modell der Garchinger Forscher übernehmen elektrisch neutrale Atome im optischen Gitter die Rolle der Elektronen. Die Atome können sich von Mulde zu Mulde im „Eierkarton“ ähnlich frei bewegen, wie die Elektronen in der Hülle eines realen Moleküls. Die große Hürde dabei: Elektronen stoßen sich wegen ihrer gleichnamigen elektrischen Ladung voneinander ab. Diese so genannte „Coulomb-Wechselwirkung“ wirkt bis über weite Entfernungen. Die Atome im „Eierkarton“ wechselwirken aber nur mit ihrem direkten Nachbarn. „Zusätzlich müssen wir also auch die charakteristische Abnahme der Coulomb-Wechselwirkung mit dem Abstand der simulierten Elektronen modellieren“, so Argüello-Luengo.
Um dieses Problem zu lösen, nahmen sich die Theoretiker als Vorbild, wie die Coulomb-Wechselwirkung in der Quantentheorie beschrieben wird. Demnach gibt ein Elektron ein Lichtteilchen (Photon) ab, das ein anderes Elektron einfängt. Wie zwei Personen auf Rollschuhen, von denen eine der anderen einen Ball zuwirft, die diesen fängt. Dadurch rollen die Personen voneinander weg. Aus analogen Gründen stoßen sich die beiden Elektronen ab. Ähnlich gestalteten es die Garchinger Forscher in ihrem modellierten Molekül. Zunächst wird jede Mulde im „Eierkarton“ mit weiteren Atomen gefüllt.
Jedes dieser einzelnen Atome kann über die Strahlung des Laserlichts energetisch angeregt werden. Die Hintergrund-Atome bilden das Medium für die Übertragung der Wechselwirkung. Ein angeregtes Hintergrund-Atom reicht die Energie dann an seinen Nachbarn weiter, und dieser an den seinen und so weiter. Die Anregung bewegt sich wie ein Photon durch das Medium. „Die Anregung entsteht bevorzugt an den Orten, an denen eines der modellierten Elektronen sitzt“, erklärt Argüello-Luengo. Das „Elektron“ und das angeregte Hintergrund-Atom stoßen sich ab. Wenn die weiterreisende Anregung auf das zweite „Elektron“ trifft, wirkt die Abstoßung ebenfalls. So wird diese vermittelt. Die Wahrscheinlichkeit für so einen Austausch nimmt mit dem Abstand der beiden „Elektronen“ ab – genauso wie bei der Coulomb-Wechselwirkung.
Spannenderweise kann der von den Theoretikern vorgeschlagene Molekülsimulator auch viel größere Architekturen modellieren als Wasserstoff. Man könne die Simulationen in Zukunft etwa nutzen, um sie mit herkömmlichen Computermodellen zu vergleichen und diese entsprechend zu justieren. Der Physiker wagt einen Ausblick: „Unsere Arbeit eröffnet die Möglichkeit, die elektronischen Strukturen von Molekülen mit analoger Quantensimulation effizient zu berechnen. Dadurch können wir chemische und biochemische Probleme bedeutend besser durchdringen, als es mit heutigen Computern möglich wäre“.
Diese Newsmeldung wurde via Informationsdienst Wissenschaft erstellt.

{kind=link}